18年来美国首个阿尔茨海默病药获批,《科学》:有效性存疑
2021-08-06 21:54:36 作者:佚名
美国当地时间6月7日,渤健(Biogen)公司的阿尔兹海默症新药获美国FDA加速批准,成为自2003年以来在美国批准的第一个新的阿尔茨海默病药物。该药物为单克隆抗体aducanumab。
《科学》称,这是一项震惊业内专家的有争议的决定。美国FDA否决了一组顾问的结论。尽管来自两项大型临床试验关于该药物减缓患者认知能力下降的能力的证据薄弱且相互矛盾,但FDA还是做出了这一决定。
在宣布该决定时,FDA承认这些对早期疾病患者的研究“在临床获益方面存在不确定性”。但FDA批准的理由是:强有力的证据表明该药物能可以清除β淀粉样蛋白,这种蛋白质积聚在阿尔茨海默氏症患者的大脑中,被认为会导致神经元损伤。FDA表示,减少这些斑块“有理由预测这对患者有着重要益处”。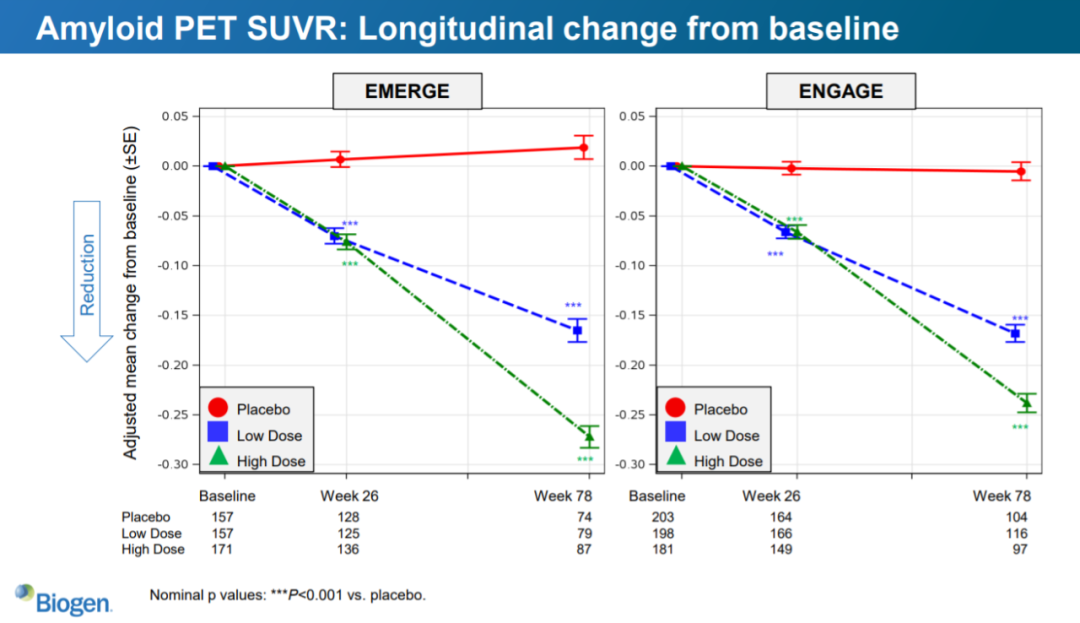
然而,近年来几种淀粉样蛋白靶向药物都未能在大型临床试验中显示出益处。FDA的决定受到了部分研究人员和患者权益团体的欢迎,但也有评论认为这是对审批标准的削弱,是一种危险的行为。
《科学》援引美国耶鲁大学医学院FDA监管政策专家约瑟夫·罗斯称,“这一批准将对FDA和美国整个医疗保健系统产生巨大的影响,”他打了一个比方:“这就像他们抽走了一根线,整个系统开始走向瓦解。”他说,FDA在最后一刻从标准批准程序转换为不需要临床改进而是依赖于“替代终点”β淀粉样蛋白的“加速途径”,这是前所未有的。他补充说,健康保险提供者和患者现在将承担未经证实的治疗费用。
阿尔茨海默病是一种进行性神经系统疾病,会损害患者的思维、记忆力和独立性,导致过早死亡。该疾病目前无法阻止、延迟或预防,正成为一个日益严重的全球健康危机,影响着疾病患者及其家人。根据世界卫生组织(WHO)的数据,全世界有数千万人患有阿尔茨海默病,这一数字在未来几年还会增长,从而使得疾病所需的医疗资源出现不足,花费将高达数十亿美元。
FDA将要求渤健完成批准后的临床试验,以验证该药物的益处。渤健之前的两次大型试验,包括来自20个国家的3000多人,结果非常模糊。2019年,渤健和共同开发aducanumab的日本制药公司卫材(Eisai)宣布,在对数据进行的中期分析表明该药物对患者没有益处后,他们将停止这两项试验。2019年晚些时候,他们又表示根据新的可用的患者数据,他们提出了新的分析。在该分析中,一项研究显示,与服用安慰剂的组相比,服用两种aducanumab剂量中较高剂量的组的认知能力下降减少了22%;第二项研究仍然没有发现任何好处。
这一逆转让许多研究人员感到困惑和怀疑。一些人敦促美国FDA在批准前要求进行另一项大型试验,认为渤健没有证明潜在的好处超过了副作用的风险,包括脑肿胀等。在FDA于2020年11月召开的独立咨询委员会会议上,专家们以压倒性多数投票认为阳性试验的数据不是该药物“有效性的主要证据”。当时没有讨论基于淀粉样蛋白减少的加速批准的可能性。
FDA很少做出违背其顾问的建议。在对2008年至2015年FDA裁决的分析中,螺丝及其同事发现89%的药物批准决定与咨询委员会的建议一致。在那些没有这样做的情况下,77%的裁决涉及比顾问建议的更严格的裁决——选择拒绝委员会投票批准的药物。在致FDA咨询委员会的一封信中,FDA药物评估和研究中心神经科学办公室主任比利·邓恩(Billy Dunn)指出,阿尔茨海默氏症治疗存在“巨大的未满足医疗需求”,以证明转向不同的批准标准是合理的。
通过破坏淀粉样斑块来对抗认知能力下降一直是阿尔茨海默氏症药物开发的主要策略。美国国家老龄化研究所所长理查德·霍德斯(Richard Hodes)说,这一决定“当然提供了继续这一研究路线的理由”。然而,他指出治疗阿尔茨海默氏症的策略正在迅速多样化,而不仅仅是淀粉样蛋白。
其他淀粉样蛋白靶向药物正在研发中。由礼来公司开发的一种抗体药物donanemab在3月份发表在《新英格兰医学杂志》上的一项2期试验中显示出有益的迹象。礼来目前正在扩大一项正在进行的试验,希望能在2023年确认这些结果并支持FDA的批准。(本文来自澎湃新闻,更多原创资讯请下载“澎湃新闻”APP)
《科学》称,这是一项震惊业内专家的有争议的决定。美国FDA否决了一组顾问的结论。尽管来自两项大型临床试验关于该药物减缓患者认知能力下降的能力的证据薄弱且相互矛盾,但FDA还是做出了这一决定。
在宣布该决定时,FDA承认这些对早期疾病患者的研究“在临床获益方面存在不确定性”。但FDA批准的理由是:强有力的证据表明该药物能可以清除β淀粉样蛋白,这种蛋白质积聚在阿尔茨海默氏症患者的大脑中,被认为会导致神经元损伤。FDA表示,减少这些斑块“有理由预测这对患者有着重要益处”。
然而,近年来几种淀粉样蛋白靶向药物都未能在大型临床试验中显示出益处。FDA的决定受到了部分研究人员和患者权益团体的欢迎,但也有评论认为这是对审批标准的削弱,是一种危险的行为。
《科学》援引美国耶鲁大学医学院FDA监管政策专家约瑟夫·罗斯称,“这一批准将对FDA和美国整个医疗保健系统产生巨大的影响,”他打了一个比方:“这就像他们抽走了一根线,整个系统开始走向瓦解。”他说,FDA在最后一刻从标准批准程序转换为不需要临床改进而是依赖于“替代终点”β淀粉样蛋白的“加速途径”,这是前所未有的。他补充说,健康保险提供者和患者现在将承担未经证实的治疗费用。
阿尔茨海默病是一种进行性神经系统疾病,会损害患者的思维、记忆力和独立性,导致过早死亡。该疾病目前无法阻止、延迟或预防,正成为一个日益严重的全球健康危机,影响着疾病患者及其家人。根据世界卫生组织(WHO)的数据,全世界有数千万人患有阿尔茨海默病,这一数字在未来几年还会增长,从而使得疾病所需的医疗资源出现不足,花费将高达数十亿美元。
FDA将要求渤健完成批准后的临床试验,以验证该药物的益处。渤健之前的两次大型试验,包括来自20个国家的3000多人,结果非常模糊。2019年,渤健和共同开发aducanumab的日本制药公司卫材(Eisai)宣布,在对数据进行的中期分析表明该药物对患者没有益处后,他们将停止这两项试验。2019年晚些时候,他们又表示根据新的可用的患者数据,他们提出了新的分析。在该分析中,一项研究显示,与服用安慰剂的组相比,服用两种aducanumab剂量中较高剂量的组的认知能力下降减少了22%;第二项研究仍然没有发现任何好处。
这一逆转让许多研究人员感到困惑和怀疑。一些人敦促美国FDA在批准前要求进行另一项大型试验,认为渤健没有证明潜在的好处超过了副作用的风险,包括脑肿胀等。在FDA于2020年11月召开的独立咨询委员会会议上,专家们以压倒性多数投票认为阳性试验的数据不是该药物“有效性的主要证据”。当时没有讨论基于淀粉样蛋白减少的加速批准的可能性。
FDA很少做出违背其顾问的建议。在对2008年至2015年FDA裁决的分析中,螺丝及其同事发现89%的药物批准决定与咨询委员会的建议一致。在那些没有这样做的情况下,77%的裁决涉及比顾问建议的更严格的裁决——选择拒绝委员会投票批准的药物。在致FDA咨询委员会的一封信中,FDA药物评估和研究中心神经科学办公室主任比利·邓恩(Billy Dunn)指出,阿尔茨海默氏症治疗存在“巨大的未满足医疗需求”,以证明转向不同的批准标准是合理的。
通过破坏淀粉样斑块来对抗认知能力下降一直是阿尔茨海默氏症药物开发的主要策略。美国国家老龄化研究所所长理查德·霍德斯(Richard Hodes)说,这一决定“当然提供了继续这一研究路线的理由”。然而,他指出治疗阿尔茨海默氏症的策略正在迅速多样化,而不仅仅是淀粉样蛋白。
其他淀粉样蛋白靶向药物正在研发中。由礼来公司开发的一种抗体药物donanemab在3月份发表在《新英格兰医学杂志》上的一项2期试验中显示出有益的迹象。礼来目前正在扩大一项正在进行的试验,希望能在2023年确认这些结果并支持FDA的批准。(本文来自澎湃新闻,更多原创资讯请下载“澎湃新闻”APP)











