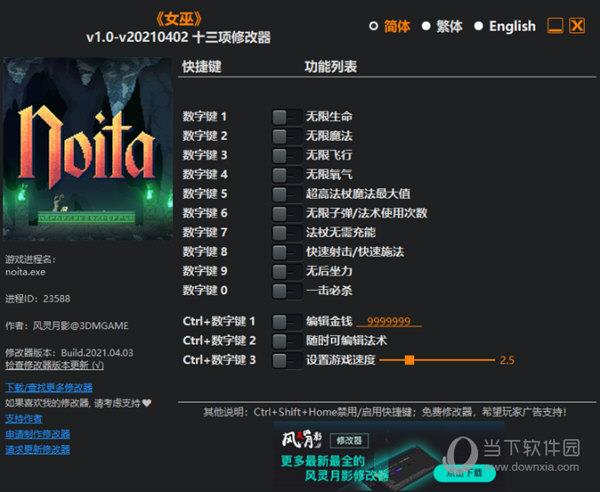
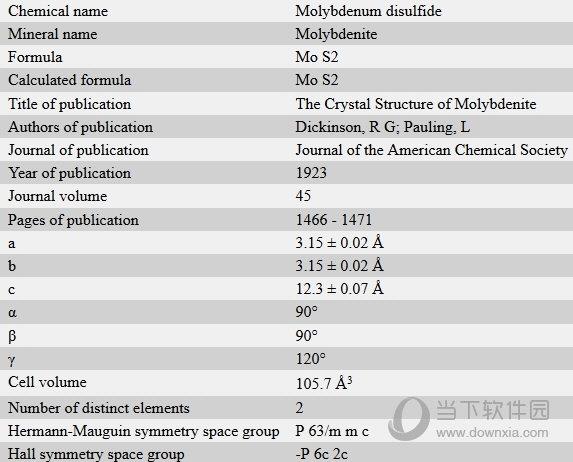
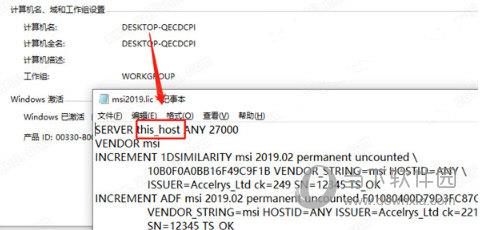
materials studio2019是一款功能十分强大的材料科学仿真模拟软件,该软件专门为从事该领域的科学家们量身定制,能够为用户提供专业的三维结构建模方案,软件结合强大的现代工业技术,帮助用户对各种晶体、无定型以及高分子材料的性质及相关过程进行深入的研究,更好地了解原子与分子的层级结构,大大提高用户的研发效率,有需要的小伙伴千万不要错过哦。
【功能介绍】
1、加入了新的反应动力学 KMC 模块 Kinetix
2、Cantera 加入反应敏感性分析和升温脱附功能
3、MS2019 中 MKL 库 和 Intel 编译器的升级提升量化计算效率
4、CASTEP 中采用基于 OPENMP/MPI 的方法减少内存使用
5、DMol? 中引入 SCAN(Meta-GGA)交换相关函数
6、DFTB+ 中加入用于电解液模拟设计 LIB2019 材料参数库
7、GULP 中新增专门针对 Li-Mn-O 正极材料的 MEAM-2NN-Qeq 力场
8、ONETEP 用户界面新增输运功能以及 TD-DFT 光学性质计算
【软件特色】
1、加速创新
与单独的测试和实验相比,该软件使材料科学家和研究团队能够更快,更高效地开发新的,性能更好,成本更高的材料。
2、降低成本
引入新材料所需的实验数量减少了10倍。
3、提高效率
通过创建可重用的建模和模拟协议,自动执行重复或繁琐的建模任务。
4、协作
捕获和分享专家知识和方法,使计算科学在组织和地理范围内更加一致。
5、解决您最棘手的问题
BIOVIA的专家科学家团队确保及时提供支持和专业知识,帮助解决材料科学中最具挑战性的问题。
【模块功能】
反应动力学模块更新
新模块:Kinetix 模块
模块的特点:
1、晶体表面的化学物理过程模拟
2、动力学蒙特卡罗方法
3、具备原子尺度的空间分辨率
4、可模拟的时间尺度可与真实实验相当
模块的应用:
a) 催化剂表面的化学反应
b) 温度递升脱附
c) 表面有序化的模拟
d) 电池电极表面的反应
Cantera 模块
1) Cantera 模块的 CSTR(连续搅拌釜反应器)计算任务中加入了敏感性分析。此项新功能使用户能够判断出反应链中最关键的步骤,减少不重要的副反应,为复杂的具有多步反应机理的模拟节省时间。
2) 新增一个能够模拟程序升温脱附 (Temperature Programmed Desorption) 的功能
3) 新增在 Cantera 中引入第三方反应机理的功能
量子力学模块更新
CASTEP 模块
1) 新增预调制的 LBFGS 结构优化方法
2) 有限位移方法计算声子散射谱和态密度的效率提高
3) 新的超胞选取方法以及插值方法的引入加速了力常数矩阵的求解
4) DFT+U 的方法扩展至固体核磁(NMR)的化学位移计算
对于顺磁性体系可以选择 spin polarize 的计算
DFT+U 不适用于 G-tensor 和 J-coupling 的计算
拓展了 CASTEP NMR 可以处理的体系范围,明显提升含有稀土元素体系的 NMR计算精度
5) 新增多体色散修正方法 (MBD@rsSCS) 处理长程弱相互作用
以 TS 为基础发展而来,但能更好地描述弱相互作用系统。可以计算原子受力和应力;
相对于 Grimme 、OBS 以及 TS 等双体修正方法,具有更好的通用性,更高的精度;
支持包括 PBE、PBE0、HSE03 以及 HSE06 等交换相关泛函。
色散修正支持所有的几何优化和性质计算。
6) 引入 Hybrid OpenMP/MPI 并行方法,减少内存占用
7) SOC 的计算结果可进行密立根布局分析
8) CASTEP 中有限位移计算振动性质时加入了“内插法”的选项
Dmol3 模块
1) 新增 SCAN meta-GGA 泛函
能够预测包括各种化学键:共价键、金属键、离子键、氢键以及范德瓦尔斯键的分子以及材料的几何结构和能量,还更好的体现了中程的范德华相互作用,在预测冰和水分子团结构,半导体材料以及二维材料的结构和电子态研究中能得到更好的结果
泛函在计算各种固体的各种性质(尤其是能量相关性质)中比 LDA 和 GGA 有很大的改进,几乎达到了杂化泛函的水平,但是比杂化泛函要大大节约时间
SCAN 泛函在内聚能、形成能的计算中普遍优于其他的半局域泛函,例如在硅的间隙缺陷形成能计算中得到与实验一致的结果
2) 在几何优化与过渡态优化计算面板中允许通过笛卡尔坐标进行结构优化
3) 在计算结果文件 outmol 中简化了交换相关泛函的描述并使得结果的呈现对于用户更加的友好
4) 几何优化以及过渡态优化过程的结果报告中更好地解释了达到能量收敛标准后优化过程仍然继续执行的原因,提高了结构优化效率
DFTB+ 模块
1) 新增专门用于锂离子电池中电解液模拟设计的 Slater-Koster Library 材料参数库 LIB2019。库中包含了 Li,C,H,O,N,F,P元素的相应参数,并且能够衡量锂离子在电解质中的扩散性能
15647151925261357qAX.png
图4 模拟典型锂离子电池电解质碳酸亚乙酯(EC)的分解
2) 新增完整的参数化脚本工作包 (DFTB+ Parameterizarion Tool Set):其中包含有四个工具帮助用户更好地制作 DFTB+参数
DFTB+ Generate Electronic Parameters
DFTB+ Electronic Evaluation
DFTB+ Final Evaluation
DFTB+ Merge Parameters
(该脚本工具包已被加入到了 BIOVIA Mateirials Studio Community 中,用户自行注册之后便可搜索关键词 DFTB Parameterization 进行下载。)
ONETEP 模块
1) 支持 Perl Script 调用
2) 在计算参数界面中添加设置所有种类元素的全局 NGWFs(non-orthogonal generalized Wannier functions) 半径的选项
3) 新增界面输运性能计算功能
4) 新增 TD-DFT 计算界面
5) 新增界面 LDOS 计算
6) 新增金属自旋的优化
7) 新增 Hybrid OpenMP/MPI 的使用
分子力学及动力学模块更新
GULP 模块
1) 新增针对含能材料的反应力场 ReaxFF RDX
2) 扩展了 MEAM-2NN 力场所适用的元素范围 (包含Cu, Ag, Au, Ni, Pd, Pt, Al, Pb, Fe, Cr, Mo, W, V, Nb, Ta, C, Si, Ge, In, Mg, Ti, Li, Ga, N, O, H, Mn, Ca,Co, Mn, P, Sn, Y, Zr)
3) 新增用于模拟 Li-Mn-O 体系(锂离子电池正极材料)的 MEAM-2NN-Qeq 力场
4) 添加了新的立场形式:Slater势和 Buffered 14-7 Lennard-Jones 势
5) 新增 ReaxFF and MEAM 力场对于振动性能计算的支持
6) 嵌入了第三方软件 Alamode,可通过玻耳兹曼输运方程计算热导率
Forcite Plus模块
1) 新增将xsd文件转化为 Abaqus 支持的 inp文件
文件中必须包含各种组分的密度场数据;
可直接通过菜单 export 为 Abaqus 支持 inp文件;
长度基本单位会由埃转化为微米;
在每个点上密度值最大的材料组分被定义为 Abaqus 模型中该格点所代表的材料
2) 新增 Tabulated torsion potential
3) 新增对带电体系施加电场的功能
其他更新
Visualizer 模块
1) 轨迹动画输出成 AVI文件时默认进行压缩
2) 可以通过 Pipeline Pilot 的 protocol 通过网页查看计算文件目录
3) 使用了一种新算法来预览三维周期性体系反应过程
 V2019 官方版")